Sindromul Pfeiffer: cauze, simptome, tratament
Sindromul Pfayffer este extrem de raro boală genetică care apare în medie la unul din 100.000 de nou-născuți. La fel de des afectează și băieții și fetele. Principalul simptom al bolii este coalescența timpurie a oaselor craniului în procesul de embriogeneză, din cauza căreia creierul nu mai poate evolua în mod normal.
Informații generale
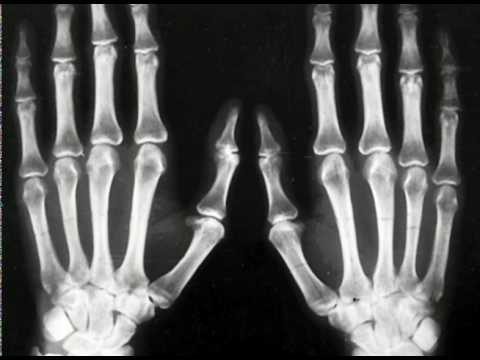
Boala a fost descoperită de faimosul germangeneticianul Rudolf Pfeiffer. În 1964, el a descris simptomele unei boli necunoscute anterior, principalele semne ale cărora au fost anomalii în structura craniului și fuziunea degetelor pe picioare sau pe mâini (syndactyly). Sindromul a fost observat la opt pacienți din aceeași familie, ceea ce a indicat natura genetică a bolii. Studii ulterioare au arătat că sindromul Pfeiffer este transmis printr-un tip dominant autosomal.
Ecografia poate detecta această boală la făt deja în cel de-al doilea trimestru de sarcină. La ultrasunete, se observă anomalii ale dezvoltării craniului, organelor interne și a extremităților.
simptome
În 1993, geneticianul american Michael CohenEl a propus clasificarea sindromului Pfeiffer în trei tipuri, în funcție de gravitatea simptomelor. Acum, clasificarea sa este în general acceptată și utilizată pe scară largă în literatura medicală.
Simptomele sindromului Pfayffer de tip 1 includcraniosynostosis - o afectiune in care una sau mai multe suturi craniene prematur fuzionează pentru a forma un os integrantă. Ca urmare, există schimbări în forma craniului și caracteristici faciale anormale - hipertelorism, urechi set-joase. Este posibil să apară o deteriorare a vederii, o presiune intracraniană crescută, un palat intestinal. La 50% dintre pacienți, se observă pierderea auzului. Cei mai mulți oameni cu acest tip de boala au niveluri normale de inteligență și nu au anomalii neurologice.
La copiii cu sindrom Pfayffer de tip 1, externsemne, cum ar fi degetele scurte pe brațe și picioare, sunt observate aproape întotdeauna. Syndactyly este un simptom posibil, dar nu necesar, pentru acest tip de patologie.
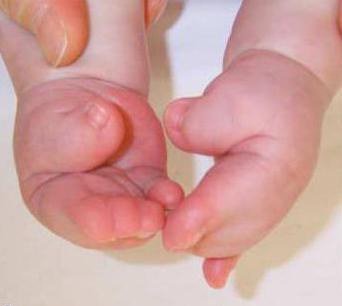
Acest tip de sindrom este moștenit autosomal dominant.
Mai jos este o fotografie a sindromului Pfeiffer. Speranța de viață în Rusia pentru persoanele care suferă de această boală depășește 60 de ani.

Pentru boala de tip 2, craniul este caracterizat de aforma „trefoil“, o fuziune a vertebrelor, sindactilie si deplasarea inainte glob ocular (ptoza). Din primele zile de viață se manifestă tulburări neurologice grave.
Al treilea tip de boală este caracterizat chiar mai multcraniosinostoza timpurie. Craniul este anormal alungit și nu formează un "trefoil". Există anomalii ale dinților, hiperteleism, retard mental, malformații ale organelor interne.
Durata de viață la tipurile 2 și 3boala variază de la câteva zile la câteva luni - defectele organelor interne și tulburările neurologice nu sunt compatibile cu viața. Mutațiile responsabile pentru aceste două tipuri de sindrom Pfeiffer apar sporadic și nu sunt moștenite genetic.
motive
Sindromul Pfayffer este asociat cu mutații ale receptorului 1factor de creștere a fibroblastelor (FGFR1) pe cromozomul 8 sau de creștere a fibroblastelor receptorului factorului de 2 (FGFR2) pe cromozomul 10. Genele care afectează mutația responsabilă pentru creșterea normală a fibroblastelor, care joacă un rol-cheie în dezvoltarea oaselor organismului.

tratament
Medicina moderna nu permite inca eliminareamutații în genomul uman, chiar dacă se știe în ce gene există perestroika. Prin urmare, pentru sindromul Pfeiffer, ca și în cazul majorității altor boli genetice, tratamentul este simptomatic. Pentru pacienții cu tip 2 și 3, prognosticul este nefavorabil: numeroasele anomalii de dezvoltare nu pot fi corectate, fie din punct de vedere medical, fie din punct de vedere chirurgical.
Sindromul Pfayffer de tip 1 este compatibil cu viața. Principalul simptom al bolii - fuziunea prematură a oaselor craniului - poate fi corectată chirurgical. Operația este recomandată copiilor până la vârsta de trei luni.

În plus, intervenția chirurgicală este eficientă în cazul sindicalității simple. Tratamentul este cel mai bine pentru a începe la vârsta de 18-24 luni, în timpul creșterii active a degetelor și degetele de la picioare.